Rishi P. Singh, MD; Robert L. Avery, MD; Mark R. Barakat, MD; Judy E. Kim, MD; Szilárd Kiss, MD
Age-related macular degeneration (AMD) is the leading cause of severe visual impairment and irreversible blindness in industrialized countries.1 In 2019, it was estimated that in the United States (US), there were 18.34 million people living with early-stage AMD and an additional 1.49 million with late-stage AMD.2 Prior to 2005, clinicians had limited options to manage the neovascular form of AMD (nAMD) and prevent progressive blindness in these patients. Over the past two decades, the most pivotal advancement in the management of retinal diseases, including nAMD, has been the implementation of vascular endothelial growth factor (VEGF)inhibitors. Anti-VEGF therapies, most of which target VEGF-A, have been fundamental in reducing legal blindness in Westernized countries by an estimated 50% to 70%.3 Intravitreal injections of anti-VEGF agents, administered at regular intervals remain the preferred treatment for nAMD to maintain adequate disease control and retain or improve visual acuity or slow down the loss of vision. Based on Current Procedural Terminology code (67028) entries for Medicare Part B, US ophthalmologists administered more than 3.5 million intravitreal injections in 2020.4
Currently approved anti-VEGF agents indicated for the treatment of retinal diseases include pegaptanib, ranibizumab, aflibercept, brolucizumab, and faricimab, as well as biosimilars ranibizumab-nuna and ranibizumab-eqrn. Additionally, bevacizumab, the first anti-VEGF monoclonal antibody to receive US Food and Drug Administration (FDA) approval in 2004, albeit for colorectal cancer, has been widely used off label to treat retinal diseases. Berkowitz et al. reported that in 2015, 1,147,432 intravitreal bevacizumab injections were administered, compared to agents approved for retina indications, aflibercept and ranibizumab (mean of 0.3-mg and 0.5-mg doses) with 870,843 and 697,412, respectively.5
In this narrative review, we summarize the evidence and discuss the role of off-label bevacizumab in the treatment and management of retinal diseases, its mechanism of action, current challenges, and provide a critical appraisal of current evidence and clinical implications.
BEVACIZUMAB MECHANISM OF ACTION
Bevacizumab is a recombinant humanized monoclonal IgG1 that binds to and inhibits the receptor-binding domain of all VEGF-A isoforms. This binding prevents interaction between VEGF-A and its receptors (Flt-1 and KDR) on the surface of endothelial cells, which initiates the intracellular signaling pathway that leads to endothelial cell proliferation, new blood vessel formation, and vascular leakage.
HISTORY OF BEVACIZUMAB IN NAMD
Philip J. Rosenfeld, MD, PhD, of Bascom Palmer Eye Institute in Miami can be credited with the introduction of bevacizumab into clinical retina practice. He had been an investigator in the early-phase trials of intravitreal injections of ranibizumab in 2001 for Genentech and witnessed the profound beneficial effect of anti-VEGF in eyes with nAMD when evaluated by optical coherence tomography (OCT).6,7 Ranibizumab, which is an antibody fragment, was derived from the same nucleic acid sequences as bevacizumab, but genetically engineered and affinity matured into a higher affinity molecule, and that early primate studies had demonstrated the molecular progenitor for bevacizumab effectively prevented iris neovascularization.8 Both ranibizumab and bevacizumab had the same anti-VEGF binding domains and would be expected to deliver near-identical anti-VEGF activity. Genentech did not wish to pursue clinical trials of bevacizumab for nAMD, so a donation-funded trial of systemically delivered (intravenously) bevacizumab was initiated at Bascom Palmer in 2004, led by Dr. Rosenfeld, in collaboration with Andrew Moshfeghi, MD, and Department Chair Carmen Puliafito, MD.
Eighteen patients received 5 mg/kg systemic infusions of bevacizumab (~500x eventual intravitreal dose) at two-week intervals and demonstrated the same dramatic improvements on OCT and visual acuity observed in the intravitreally administered ranibizumab trial. However, the systemic dosing of bevacizumab raised potential safety concerns associated with systemic hypertension and thromboembolic events.
The commercial concentrations of both bevacizumab (25 mg/mL) and ranibizumab (10 mg/mL) were considered, and it was recognized that the molecular weight of bevacizumab was approximately three times that of ranibizumab, and thus the molarity was nearly identical, meaning that equal volume of bevacizumab would contain a nearly equal number of molecules as ranibizumab. In May 2005, the first intravitreal injection of bevacizumab, dispensed into individual syringes, was administered as salvage therapy to a patient at Bascom Palmer who had been responding poorly to intravitreal pegaptanib therapy. Within one week of an approximately 1.0-mg intravitreal dosage of bevacizumab, resolution of subretinal fluid was observed on OCT, and visual acuity remained stable for four weeks with no sign of inflammation.9 This initial case report generated excitement within the retina community of a potentially effective and safe treatment option for patients with nAMD, eventually leading to wider adoption.
Early in the evolution of nAMD treatment with intravitreal bevacizumab, Dr. Rosenfeld and Serafin Gonzalez, PharmD, CPh, BCSCP, Director of the Department of Pharmacy at Bascom Palmer, developed the sterile technique for safely dispensing and aliquoting the bevacizumab from the vial into individual syringes and stressed the importance of only a single entry into the source vial. They ensured that this dispensing technique was made widely available.
Nearly 20 years later, bevacizumab is still among the most used anti-VEGF agents and has remained in the armamentarium of retina specialists, globally.
CLINICAL TRIALS OF BEVACIZUMAB IN NAMD
Despite the off-label status of intravitreal bevacizumab for nAMD, the benefits of its use in the treatment of nAMD have been demonstrated in multiple clinical trials (Table 1). The CATT (Comparison of Age-Related Macular Degeneration Treatments Trials) trial was the first large, multicenter noninferiority trial of bevacizumab and ranibizumab for nAMD.10 The dosages evaluated were 0.50 mg (in 0.05 mL of solution) for ranibizumab and 1.25 mg (in 0.05 mL of solution) for bevacizumab. It is important to note that administered bevacizumab was repackaged in glass vials in a Good Manufacturing Practices (GMP)-compliant aseptic filling facility, under a US FDA investigational new drug application. A total of 1,208 patients were randomized, and results demonstrated that with respect to visual acuity, bevacizumab administered monthly was noninferior to ranibizumab administered monthly, with 8.0 and 8.5 letters gained, respectively. Similarly, bevacizumab, administered as needed with monthly evaluation was equivalent to ranibizumab as needed at 1 year, with 5.9 and 6.8 letters gained, respectively. Both drugs resulted in a substantial reduction in total retinal thickness at the fovea after the first injection when evaluated at four weeks, with no fluid seen on OCT for 27.5% of eyes treated with ranibizumab and 17.3% treated with bevacizumab. However, results at one year favored ranibizumab, with ranibizumab-treated eyes being more likely to have no fluid on OCT when compared with bevacizumab (43.7% versus 26.0%; P < 0.001). Rates of death, myocardial infarction, and stroke were similar for patients receiving either bevacizumab or ranibizumab. Overall, the proportion of patients with serious systemic adverse events (primarily hospitalizations) was higher with bevacizumab than with ranibizumab (24.1% versus 19.0%) for a broad distribution of events in disease categories not identified in previous studies as areas of concern.10
Similarly, the IVAN trial also compared the efficacy and safety of intravitreal ranibizumab and bevacizumab to treat nAMD at fixed monthly and as needed dosing regimens.11 At one year, the visual acuity changes were equivalent, and there were no differences in total foveal thickness between the two anti-VEGF agents for both dosing regimens. The authors concluded that the drugs and treatment regimens demonstrated similar efficacy and safety.
Investigators in France conducted the multicenter, noninferiority, double-masked, randomized GEFAL clinical trial.12 The study included 501 patients that were randomly assigned to intravitreal bevacizumab (1.25 mg) or ranibizumab (0.50 mg). Hospital pharmacies were responsible for preparing, masking, and dispensing treatments. At one year, based on an as-needed dosing regimen, the mean number of injections was 6.8 in the bevacizumab group and 6.5 in the ranibizumab group, and bevacizumab was noninferior to ranibizumab in visual acuity (+1.89 letter difference). Both drugs reduced the central subfield macular thickness, with a mean decrease of 95 μm for bevacizumab and 107 μm for ranibizumab, and the proportion of patients with serious adverse events (SAEs) was similar (12.6% for bevacizumab and 12.1% for ranibizumab). The authors also noted that comparative studies in the literature need to have a greater focus on the importance of drug preparation and include descriptions of how the 4-mL or 16-mL preservative-free, single-use vials were aliquoted into smaller doses for intravitreal use. They emphasized that the repackaging must follow meticulous sterile technique and be done under a positive-pressure sterile hood.12
The LUCAS trial compared the efficacy and safety of bevacizumab versus ranibizumab when administered according to a treat-and-extend regimen.13 Of the 441 patients treated at 10 centers in Norway, at one year, bevacizumab was equivalent to ranibizumab, with 7.9 and 8.2 mean letters gained, respectively. There was no significant difference in central retinal thickness, with a mean decrease of 112 μm for bevacizumab and 120 μm for ranibizumab. Although not powered to demonstrate statistical differences in safety, there were fewer arteriothrombotic events in the bevacizumab group (1.4%) than in the ranibizumab group (4.5%) and significantly more cardiac events in the ranibizumab group.13
The BRAMD study was a multicenter, randomized, controlled, double-masked clinical trial of 327 patients with nAMD treated monthly with 1.25 mg bevacizumab or 0.5 mg ranibizumab in The Netherlands.14 The mean gain in visual acuity at one year was 5.1 letters in the bevacizumab group and 6.4 letters in the ranibizumab group. There were no significant differences in absolute central retinal thickness or change in central retinal thickness, and the occurrences of SAEs and adverse events were similar for both groups. The results of this study provided the foundation for official guidelines from The Dutch Ophthalmological Society that advised ophthalmologists to use bevacizumab as the preferred drug to start anti-VEGF treatment in nAMD.15
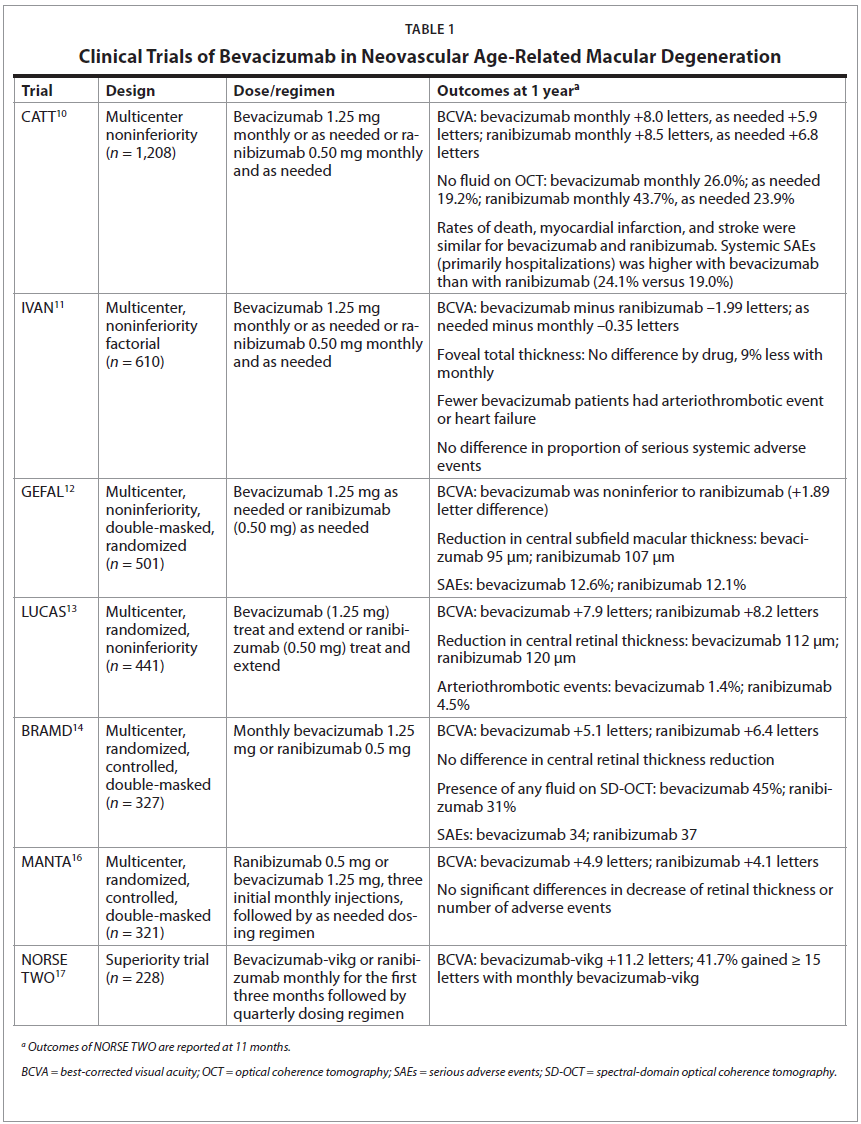
The MANTA study was a multicenter trial conducted at 10 Austrian centers.16 The study included 321 treatment-naïve patients that received either 0.5 mg ranibizumab or 1.25 mg bevacizumab. Both groups received three initial monthly injections, followed by retreatment on an as-needed dosing regimen. Results demonstrated that, at one year, the increase in visual acuity was 4.9 letters in the bevacizumab group and 4.1 letters in the ranibizumab group. Additionally, there were no significant differences in the decrease of retinal thickness, change of lesion size, or number of adverse events.
More recently, the NORSE TWO trial evaluated a formulation of bevacizumab (bevacizumab-vikg) developed specifically for the treatment of retinal conditions.17 NORSE TWO was a superiority trial, comparing the new bevacizumab formulation to ranibizumab in 228 patients with nAMD. Participants received either monthly bevacizumab-vikg or ranibizumab monthly for the first three months followed by a quarterly dosing regimen. Results demonstrated that 41.7% of eyes gained ≥ 15 letters of vision with monthly bevacizumab-vikg.
Overall, bevacizumab has consistently demonstrated equivalent visual acuity results when compared with ranibizumab, although anatomical outcomes in some trials have not been equivalent, often favoring ranibizumab. Most studies have not observed any differences in the safety profiles of the drugs, however in CATT, the proportion of patients with one or more systemic SAEs at one year was higher with bevacizumab (24.1%) compared to ranibizumab (19.0%), and at two years, the incidence of SAEs increased to 40% for patients receiving bevacizumab and 32% for patients receiving ranibizumab.10,18 A 2017 meta-analysis of individual patient data conducted by Maguire et al. aimed to estimate the relative risk of systemic SAEs and selected specific SAEs adjusted for prognostic baseline variables.19 The analysis included data from 3,052 patients that participated in five clinical trials. The authors concluded there were no large differences in risk of systemic SAEs between bevacizumab and ranibizumab. Similarly, meta-analyses conducted by Yin et al. in 2022 and Nguyen et al. in 2018 found no significant differences in the risk of death and arteriothrombotic events between bevacizumab and ranibizumab at one and two years.20,21
Despite the important recognized clinical benefits, anti-VEGF drugs for the treatment of retinal diseases are costly. Globally, in 2020, the anti-VEGF market for retinal diseases exceeded $13.1B USD.22 In 2019, the US average sales prices per dose of aflibercept and ranibizumab were $1,877 USD and $1,717 USD, respectively, as compared to approximately $70 USD for bevacizumab. Given this cost differential and the demonstrated noninferior visual acuity efficacy and safety profile, off-label bevacizumab has become the documented first-line standard of care at many US medical centers, by many insurers, and in several European countries.23 Interestingly, in 2018, members of the American Society of Retina Specialists were asked what their first-line choice of anti-VEGF for nAMD was, and 66.3% used bevacizumab, followed by 25.4% selecting aflibercept and 8.3% using ranibizumab.24
DISPENSING AND REPACKAGING OF INTRAVENOUS BEVACIZUMAB
As noted, the bevacizumab that is currently used off label in ophthalmic practices is the same agent formulated and packaged for oncologic use. To deliver bevacizumab intravitreally, it is commonly repackaged into individual syringes by compounding pharmacies for use in the eye, meaning the compounding pharmacies are repackaging the drug for off-label use. This differs from repackaging of a drug approved for a specific indication into a dosage form that has not received prior approval. The repackaging practice is permitted by the FDA in circumstances where a prescribed form or dosage is not commercially available to treat a patient.
The method of repackaging bevacizumab often includes aliquoting small doses from a large vial to create prefilled syringes for intravitreal injection. The solution is aseptically repackaged into multiple sterile syringes from one single-use vial for extended storage and subsequent injection. It is not uncommon for a single vial to be repackaged into up to 80 syringes. Although the repackaging process is permitted by the FDA, the resultant product does not meet the specific standards of products approved for use as ophthalmic injectables nor is the parenteral innovator solution ophthalmic standard compliant.
With each entry into a single vial, despite careful pharmacy procedures, the potential for contamination is increased. This practice may increase a patient’s risk of potentially blinding SAEs, including endophthalmitis, which has been reported following use of repackaged bevacizumab.25 Even in 2005, Drs. Rosenfeld and Gonzalez stressed the importance of allowing only a single entry into the source vial and disseminated their protocol.
Differences in the bevacizumab delivered to the patient can also occur as a result of human error or judgment. A review by Gonzalez of the events surrounding clusters of endophthalmitis following bevacizumab administration suggests the resultant vision loss was not the result of the drug or the injection technique, but rather of the compounding procedures used to prepare the bevacizumab. Noncompliance with recognized standards and poor aseptic technique were cited as the most likely causes, with recommendations for stricter adherence to United States Pharmacopeia (USP) Chapter 797 requirements.25 Gonzalez acknowledged that infectious endophthalmitis after intravitreal injections will never be totally eliminated from clinical practice but that USP Chapter 797 should always be followed when fractionating a vial of a bevacizumab to minimize the occurrence of the catastrophic consequences of a batch of syringes being contaminated. Although it may be more expensive to do, limiting each vial to only a single injection has been a strategy used during times of endophthalmitis outbreaks.
Studies have also demonstrated variability in the quality and quantity of repackaged bevacizumab.26-28 In 2015, Yanuzzi et al. evaluated microbial culture growth, endotoxin levels, and quantity and binding affinity of protein in samples of repackaged bevacizumab acquired from 11 compounding pharmacies in the US. Although no microbial contaminants or endotoxin was detected in any of the samples, 81% of the samples demonstrated lower protein concentrations than source bevacizumab acquired directly from the manufacturer (Genentech). Additionally, there were statistically significant differences in the protein concentration between 30% of samples acquired from the same pharmacy.26 A study conducted in the United Kingdom evaluated repackaged bevacizumab from five compounding pharmacies using microflow imaging to evaluate subvisible particle size, particle density, particle size distribution, protein concentration, immunoglobulin G (IgG) content, and molecular weight.27 Significant differences in subvisible particle density were observed between bevacizumab batches from the five suppliers. Notably, particle density increased with storage in repackaged pre-filled syringes, with the particle size distribution in the repackaged bevacizumab syringes found to be outside the range specified by the USP for injectable ophthalmic solutions.27
Another important consideration is the immediate-use syringes being used for long-term storage of the aliquoted repackaged bevacizumab, which may introduce additional challenges and variability in the final product for administration. Polypropylene syringes lubricated with silicone oil for subcutaneous use but used for intravitreal injection have been associated with the presence of intravitreal silicone oil droplets. It is also not uncommon for 0.5 mL insulin syringes with staked-in needles to be used in bevacizumab repackaging; however, these needles have been shown to be a main source of silicone oil contamination in repackaged bevacizumab.29 Dounce et al. recently explored the potential changes in bevacizumab characteristics that may occur with storage, by examining the particle loads that accumulate from various syringes containing anti-VEGF agents over time.30 Similar to the findings above, insulin syringes consistently exhibited very high particle counts, whereas oleamide-lubricated syringes had substantially fewer particles but exhibited appreciable increases over time (leading to visible particles). Baked-on silicone glass syringes and lubricant-free polymer syringes both exhibited low particle levels of ≥ 10 μm; however, lubricant-free syringes exhibited the lowest particle levels of ≥ 1 μm and the lowest particle levels with bevacizumab agitation. This option of lubricant/silicone-free syringes is now offered by many compounding pharmacies.
Although this commentary is focused on bevacizumab, adverse events associated with the repackaging of ophthalmic drugs is not unique to bevacizumab. In recent years, clusters of events have occurred and were determined to be associated with compounding facility practices.31 The FDA conducted 425 inspections of compounding pharmacies between 2012 and 2017 and found the vast majority to have “problematic” conditions. It would not be unexpected that, similarly, ophthalmic drugs repackaged under conditions that may not as stringently adhere to regulation standards may also have variable characteristics of the final product. As a result of increased regulation and enforcement, the market has been restricted to far fewer compounding facilities, and when a facility has been cited by the FDA, it causes shipping interruptions resulting in dramatic shortages of available repackaged ophthalmic drugs.32,33 It is also important to recognize that safety results reported in a clinical trial setting may have used bevacizumab repackaged in stringently regulated compliant aseptic facilities, and thus may not exactly reflect the safety profile of commercially available repackaged bevacizumab.
To mitigate the challenges associated with repackaged off-label bevacizumab and variability of repackaging practices, ideally, a bevacizumab formulation specific to ophthalmic use will be developed and fall under the regulatory controls of manufacturing a biological therapy for intravitreal use. Further, the availability of a prefilled, silicone-free syringe, containing a single-dose is preferred by retina specialists and would eliminate the identified risks associated with dose aliquoting. As noted, consistency of purity, potency, and safety of the final product are critical and adherence to GMP activities ensure a manufacturer can consistently control and produce products to meet the identity, strength, purity, and quality appropriate to their intended use. GMP facilities manufacturing biologics for ophthalmic delivery are regulated and inspected by the FDA twice annually to ensure practices are being followed.
CONCLUSION
Many factors should be considered in determining the best treatment strategy for patients with nAMD. Initial treatment selection and potential treatment switch should be based on physician and patient choice after careful consideration of the patient’s needs and circumstances. For the treatment of nAMD, bevacizumab has demonstrated noninferior visual acuity outcomes with a similar adverse systemic safety profile as ranibizumab, including rates of SAEs such as arteriothrombotic events. It is recognized that bevacizumab continues to have an established role in the management of retina diseases, as evidenced by its maintained presence in retina clinics, globally, even in an evolving and broader landscape of drug development. The community anticipates the addition of an on-label bevacizumab formulation that can deliver reliable efficacy, while having the safety benefits associated with GMP-compliant manufacturing practices.